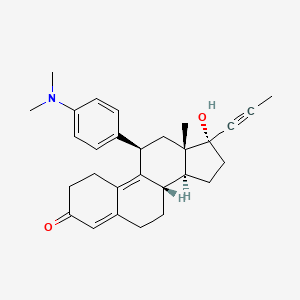
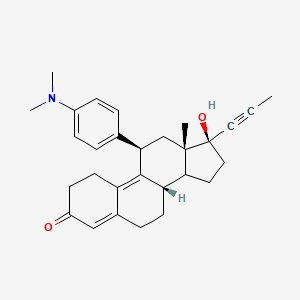
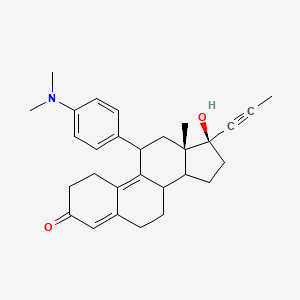
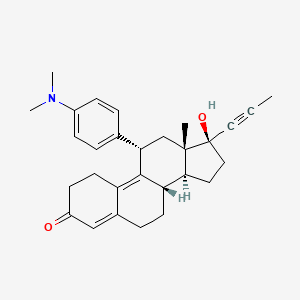
suite of automated docking tools. It is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure
AutoDock 4 uses PDBQT formatted files not only for the receptor but also for the ligand.
The PDBQT format is described here.
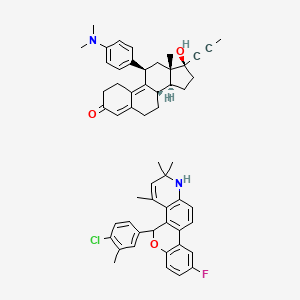
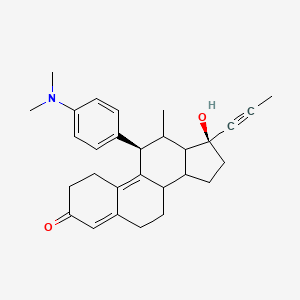
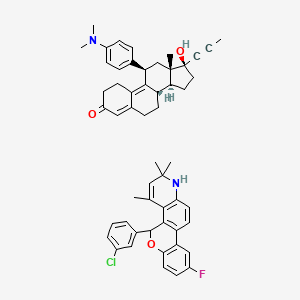
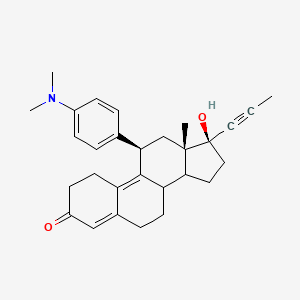
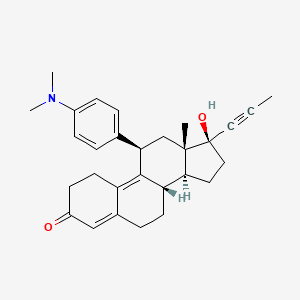
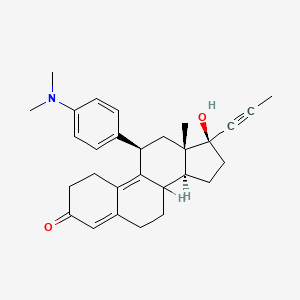
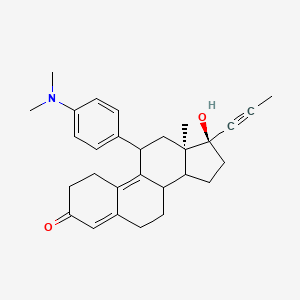
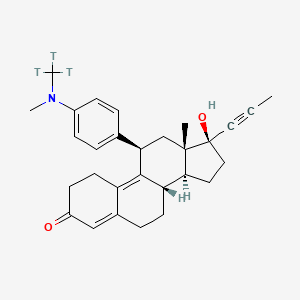
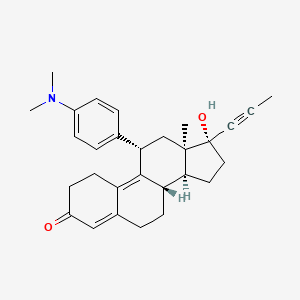
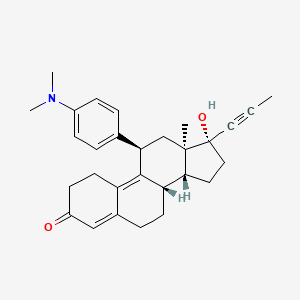
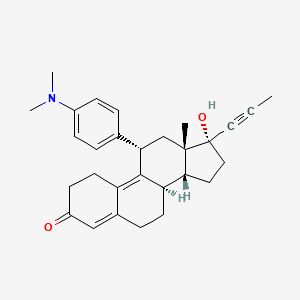
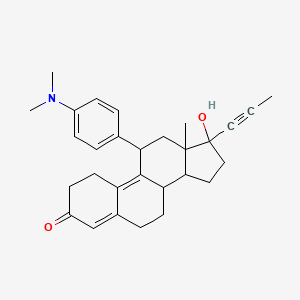
Yesterday I have converted all the MMsINC entries (about 4M) to the PDBQT format.
It is so possible to download each entry as input format for Autodock experiments.
Have a look at this example page. As you can see, at the top of the page there is a menu for the download of several format files. The first of them is Autodock input file. Simply press the "go" button to download it.
Let me close this post with this nice photo.

Where: the place is the Poetto Beach in Cagliari (Sardinia, Italy).
What: Stefano Moro and me eating these "ricci di mare" (Sea urchin).